
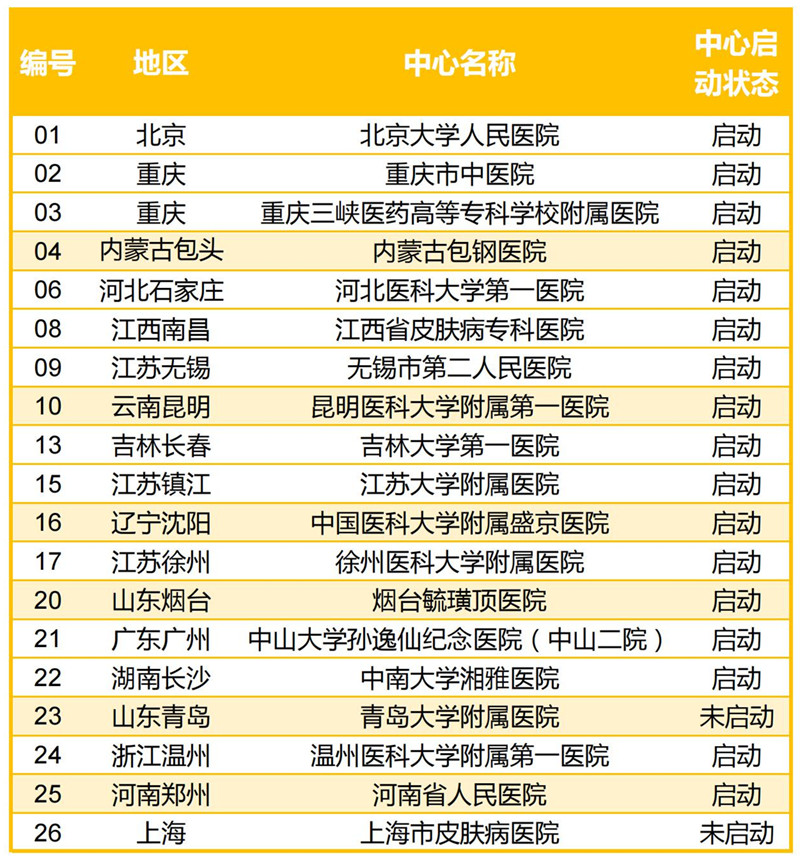
即日起,面相全国招募中重度慢性斑块状银屑病患者参与药物有效性和安全性研究临床试验,符合条件的患者可就近在北京大学人民医院、重庆市中医院等全国 26 家医疗机构参与。
该研究旨在在成年中重度慢性斑块状银屑病患者中,比较 HOT-3010 (注射剂)与修美乐®(注射剂)两组疗效的相似性以及安全性、免疫原性、药代动力学特征。
该研究已获国家药品监督管理局(NMPA)临床试验批件,并在“药物临床试验登记与信息公示平台”登记公示。
药物临床试验登记与公示

题目和背景信息
登记号:CTR20211883
相关登记号:CTR20181078
药物名称:重组全人源抗肿瘤坏死因子-α单克隆抗体注射液
药物类型:生物制品
适应症:中重度斑块状银屑病
试验专业题目:多中心、随机、双盲、平行分组评价重组全人源抗肿瘤坏死因子-α单克隆抗体注射液(HOT-3010)与修美乐®治疗成年中重度慢性斑块状银屑病的有效性和安全性研究
试验通俗题目:多中心、随机、双盲、平行分组评价 HOT-3010 与修美乐®治疗成年中重度慢性斑块状银屑病的有效性和安全性研究
方案是否为联合用药:否
(本试验信息已在NMPA“药物临床试验登记与信息公示平台”登记)
现公开招募受试者,如果您符合下列条件,将有可能入选:
1、自愿参加研究并签署书面知情同意书(ICF),且可遵守研究方案的要求接受治疗及访视。
2、筛选时年龄≥18岁且≤70岁的男性或女性。
3、中重度斑块状银屑病患者,随机前稳定病史≥6个月。中重度的定义为:银屑病面积与严重程度指数(PASI)评分≥12分,或银屑病体表受累面积(BSA)≥10%;且医生整体评价(sPGA)评分≥3分。(通俗解释:中重度斑块状银屑病的面积差不多要10个巴掌大,约为两个小腿的面积)
4、自筛选期直至最后一次给药结束后3个月内,能够采取有效避孕措施。
存在以下任何一种情况,受试者不得入选:
1)在筛选访视时,受试者患有点滴状银屑病、红皮病型银屑病、脓疱型银屑病、药物导致的银屑病、其他皮肤病变(如湿疹)、红斑狼疮或其他系统性自身免疫性炎症性病变,可能对治疗结果的评估产生影响。
2)既往接受过以下银屑病治疗:
•随机前2周内曾接受局部抗银屑病药物治疗;
•随机前4周内曾接受PUVA和/或UVB治疗、非生物药治疗(包括但不限于系统糖皮质激素、来氟米特、环磷酰胺、甲氨蝶呤、环孢素、维A酸、中药及其制剂等,其中,中药包括但不限于复方青黛丸(胶囊/片)、消银颗粒(胶囊/片)、郁金银屑片、银屑胶囊、克银丸、雷公藤多苷、复方甘草酸苷类等;中药汤剂;中药洗浴等);
•随机前4周内曾使用融合蛋白类TNF抑制剂;
•随机前12周内曾使用单抗类TNF抑制剂;
•随机前24周内曾使用IL-12/23抑制剂或IL-17抑制剂;
•随机前正在接受其他处于5个半衰期内的其他药物治疗(包括但不限于PDE-4抑制剂,JAK抑制剂等);
•既往使用TNF抑制剂或其生物类似药治疗无效或不耐受。
3)受试者有结核病史(包括结核已治愈)、活动性结核、潜伏性结核或临床表现疑似为结核感染者。
4)伴有以下活动性感染,或有以下病史:
•可能影响银屑病评价的其它活动性皮肤疾病或皮肤感染(如细菌、真菌或病毒感染);
•随机前4周内进行过全身系统抗感染治疗;
•随机前8周内患有需接受静脉抗感染治疗的严重感染;
•存在复发性、慢性或者其他活动性感染,经研究者判定参加本研究会增加受试者风险。
5)患有淋巴瘤或其他恶性肿瘤史(以下病变除外:经过彻底治疗且5年以上没有任何复发迹象的皮肤原位鳞癌、基底细胞癌、原位宫颈癌)。
6)受试者伴有活动性神经病变,包括但不限于:多发性硬化症、格林-巴利综合征(Guillian-Barre综合征)、视神经脊髓炎、横贯性脊髓炎、或有神经症状提示有中枢神经系统脱髓鞘病变者。
7)伴有中重度心力衰竭(纽约心脏病协会 [NYHA] III/IV级)的患者。
8)伴有其他严重的、进展性、或者未能控制的疾病,包括但不限于免疫系统、内分泌系统、血液系统、泌尿系统、肝胆系统、呼吸系统、精神系统、心血管系统、胃肠系统或传染性疾病,且经研究者判定参加本研究会增加受试者风险。
9)受试者对试验药物及其辅料有超敏反应,或对阿达木单抗及其辅料有过敏史。
10)随机前12周内接种过活疫苗,或有意向在研究期间及末次给药后12周内接种活疫苗。
11)筛选时实验室检查结果有以下异常:
•血红蛋白<90 g/L;
•白细胞(WBC)计数<3.5×109/L;
•血小板<100×109/L;
•总胆红素超过执行检验的实验室的正常值上限1.5倍;
•血清肌酐超过执行检验的实验室的正常值上限1.2倍;
•天门冬氨酸转氨酶(AST)或丙氨酸转氨酶(ALT)超过执行检验的实验室的正常值上限2倍;
•筛选时HBsAg、HBeAg、HCVAb、HIVAb、梅毒螺旋体抗体中任意一项阳性。HBsAg阴性、HBcAb和/或HBeAb阳性者应进一步进行HBV DNA检测,若大于等于所在研究中心实验室正常值范围上限,需排除。
•其他实验室检查结果异常,经研究者判断可能影响受试者完成试验或干扰试验结果。
12)妊娠期(育龄期女性筛选期血妊娠试验结果为阳性)及哺乳期女性患者;或试验期间及给药结束后3个月内不能采取有效避孕措施的男性及女性患者;或有捐精捐卵计划者。
13)计划在研究期间进行大手术,除非研究者评估后判定不会增加受试者风险,或者不会影响受试者接受研究药物和参加研究的依从性。
14)筛选前3个月内参加过其他临床试验且使用了试验药物者。
15)筛选前6个月内有酗酒史者(即男性每周饮酒超过28个标准单位,女性每周饮酒超过21个标准单位(1标准单位含14g酒精,如350mL啤酒、45mL酒精量为40%的烈酒或150mL葡萄酒))或者药物滥用/依赖史者;
16)既往或现有焦虑、抑郁或自杀倾向等精神疾病者;
经研究者判断,参加试验不能使受试者获益或者影响研究终点的评估。
受试者可以随时退出试验而不需要任何理由。出现以下情况时受试者可退出试验:
•受试者撤回知情同意书
•妊娠
•对研究方案的依从性差
•发生严重不良事件、实验室检查异常或其他影响疗效评价(如湿疹)的医疗状况,继续参加研究将不符合受试者的最佳获益
•不满足入组标准或满足排除标准(新出现或之前未发现的)
•在第16周时PASI自基线改善未达到50%
•研究过程中受试者服用了影响本研究疗效评价的其他药物,经研究者判断不适合继续接受研究
•其他研究者判断不适合继续接受研究的情况
受试者中止或退出研究的原因应记录在电子病例报告表(eCRF)中。提前退出研究的受试者将尽最大可能完成提前退出访视。
患者获益:
1、自入组到试验结束,共计52周,患者自愿参加,可随时退出。
2、各中心按实际访视次数给予受试者交通补贴、PK采血补贴。
研究者信息
全国共计 26 家医疗机构参与该临床研究。符合入选标准的受试者可就近参与。
如果您符合上述条件并自愿参与,报名提交后会有专业医疗人员与您取得联系。
咨询电话:400-033-0187



{kind=link}